第一作者:蒋林涛、陈兴宝
通讯作者:李蓓副教授、木士春教授
通讯单位:武汉理工大学
论文DOI:10.1021/acscatal.4c07602
全文速览
近日,武汉理工大学木士春教授和李蓓副教授课题组合作设计了一种活化羟基诱导界面水的质子交换策略,用于高效碱性析氢反应(HER)。该研究通过电化学活化方法合成了一种具有核壳结构的R-Ru-Ni(OH)2催化剂,并成功在Ru表面引入了大量活化羟基(OHad)。研究表明,OHad的引入优化了Ru活性位点的电子/能带结构和物理化学状态,并显著参与质子交换过程,改善了界面水分子的吸附和解离步骤,从而加速了催化动力学过程。具体而言,表面活化的OHad位点通过直接的OH-H2O质子交换优化了水的吸附路径,并以低至0.035 eV的吉布斯自由能实现了吸附水物种的分解;而且,额外的OHad位点改善了*H的脱附行为,打破了OH阻碍的动力学限制,并双向降低了决速步骤的能垒。在电流密度为10和100 mA cm-2碱性条件下,其HER过电位分别仅为26和81 mV,并具有出色的稳定性(在20 mA cm-2下达到100 h)。此外,作者进一步证明了金属表面活化羟基-界面水质子交换策略的普适性。本研究从实验和理论上证明了活化羟基对碱性析氢反应中界面水质子交换过程的调节,研究成果将为高效催化剂的合理设计提供指导。
背景介绍
催化剂界面关键中间体的化学状态与微观结构显著影响电化学性能。对于碱性析氢反应(HER),存在着界面水的吸附与解离以及*H、*OH中间体的形成与脱附等复杂过程,严重阻碍了HER反应的进行。一般而言,碱性HER动力学过程比酸性低2-3个数量级。因此,改善碱性HER催化剂的中间体相互作用有利于调节活性位点本征活性,降低反应能垒,加快催化动力学。通常认为,在碱性HER反应路径中,*OH/OH-的形成与脱附是不可避免的。如果采用通常改善其中间体行为的策略,催化过程中将会受到因*H/*OH竞争性吸附导致的动力学阻碍,不利于进一步优化碱性HER活性。相较而言,通过利用催化剂表面吸附的羟基物种来直接参与HER过程,有望进一步改善催化反应路径,提高本征动力学。这是因为,*OH是一种电子有利的质子受体,相较于纯的原子级金属活性中心更有利于与界面水物种相互作用,从而促进水吸附过程。另一方面,由于*OH直接参与反应形成额外的OH位点,避免了OH脱附步骤,因而加速了随后的水解离与*H脱附过程。因此,*OH位点直接参与HER反应可以双向改善碱性析氢过程,有利于打破*H与*OH竞争性动力学障碍,从而极大提高本征活性。
本文亮点
1. 本文首先合成了一种核壳催化剂Ru-Ni(OH)2,然后通过原位电化学活化方式进一步合成了具有核壳结构的R-Ru-Ni(OH)2催化剂,同时实现了Ru表面活化OHad物种的引入,有效提升其碱性HER活性。系列测试亦证明了该策略的普适性。
2. 本研究从实验和理论上证明了金属表面的活化OHad与界面水发生了显著的质子交换,实现了HER电子转移路径的优化,为高性能碱性HER催化机制设计提供了一种新颖的思路。
图文解析

图1 (a) R-Ru-Ni(OH)2制备的示意图。(b, c) NF上Ru-Ni(OH)2和R-Ru-Ni(OH)2的SEM图像。(d, e) Ru-Ni(OH)2和R-Ru-Ni(OH)2的HRTEM图像。(f, g) 通过EDS映射得到的Ru-Ni(OH)2和R-Ru-Ni(OH)2相应的元素分布情况。
通过水热合成及电化学CV活化重构等过程,获得核壳催化剂R-Ru-Ni(OH)2/NF(图1a)。通过SEM和TEM证明了通过水热法成功合成了Ru-Ni(OH)2核壳纳米结构材料(图1b,c)。其中,Ru核的直径约为80-100 nm,Ni(OH)2壳层约20 nm厚度。经电化学活化获得的R-Ru-Ni(OH)2催化剂的结构没有出现明显的变化(图1d,e)。其晶格条纹主要对应于晶体Ru,Ni(OH)2则更分布在外壳层。EDS表明Ru元素主要分布在Ru-Ni(OH)2纳米球内部,而Ni、O分布在Ru-Ni(OH)2纳米球表面。这些进一步证明了Ru-Ni(OH)2核壳结构的形成(图1f, g)。
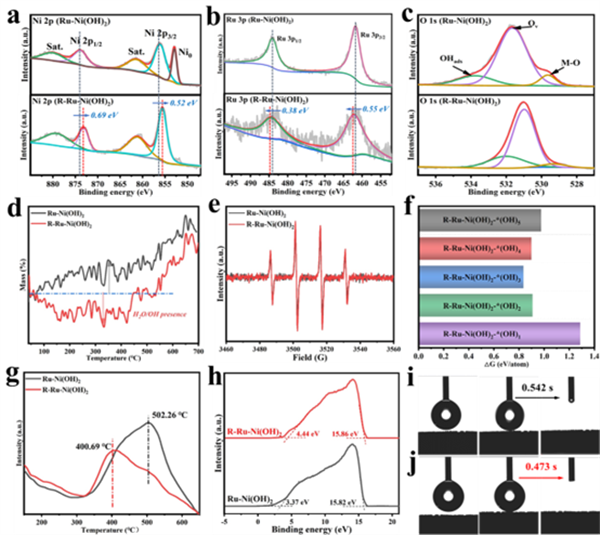
图2 (a, b, c) R-Ru-Ni(OH)2和Ru-Ni(OH)2的Ni 2p、Ru 3p和O 2p的高分辨率XPS。(d) R-Ru-Ni(OH)2和Ru-Ni(OH)2在氩气氛围下的TG曲线。(e) R-Ru-Ni(OH)2和Ru-Ni(OH)2的EPR谱图。(f) 不同数量OH吸附基团的R-Ru-Ni(OH)2-*(OH)x的形成能。(g) R-Ru-Ni(OH)2和Ru-Ni(OH)2的H2-TPD曲线。(h) R-Ru-Ni(OH)2和Ru-Ni(OH)2的UPS谱图。(i) Ru-Ni(OH)2和 (j) R-Ru-Ni(OH)2的水接触角测试。
XPS证明了催化剂经电化学活化后电子从Ru核转移到Ni(OH)2壳层上,有利于加强Ru位点对羟基基团的吸附作用(图2a,b)。同时,O 1s光谱亦证明催化剂活化后存在着更多的表面含O基团(图2c)。此外,热重曲线的下降(质量减少)验证了催化剂中存在更多的结合水与羟基物种(图2d)。从图2e可以看出,活化后的催化剂表现出更强的EPR信号,表明R-Ru-Ni(OH)2形成了更多的羟基。接着,通过DFT计算解析了Ru-Ni(OH)2中Ru催化剂表面的羟基吸附动力学,表明OHad易吸附在Ru催化剂表面(图2f)。H2-TPD证明了R-Ru-Ni(OH)2更利于H2脱附(图2g),而且由UPS分析可知,R-Ru-Ni(OH)2还有利于中间体脱附(图2h)。从图2i, j的水接触角分析可知,R-Ru-Ni(OH)2具有更优异的吸水性与亲水能力。这些研究结果均与其表面存在的亲水羟基基团有关。
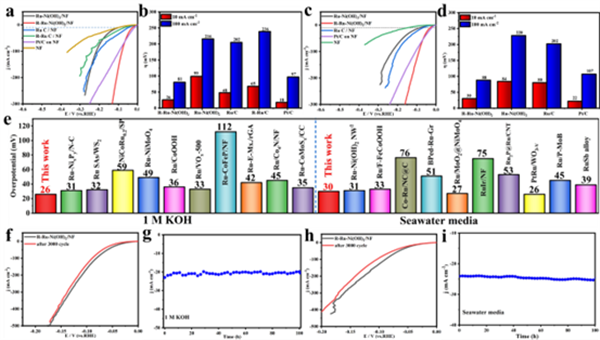
图3 催化剂在 (a, b) 1 M KOH溶液和 (c, d) 海水介质中的LSV曲线及相应过电位(@10和100 mA cm-2)。(e) 在碱性介质中,R-Ru-Ni(OH)2与近期报道的钌基电催化剂在10 mA cm-2时析氢反应(HER)性能的比较。R-Ru-Ni(OH)2在 (f, g) 1 M KOH溶液和 (h, i) 海水介质中的3000次循环伏安稳定性和电流密度随时间变化的曲线。
基于此,进一步研究了所制备催化材料在碱性条件下的析氢性能。由图3a可以看出,在所有催化剂中,R-Ru-Ni(OH)2催化剂表现出最佳的活性:在电流密度分别为10与100 mA cm-2下过电位分别为26与81 mV,可与商业Pt/C性能(18,97 mV)相媲美(图3a-e)。此外,R-Ru-Ni(OH)2亦具有优异的CV循环(3000圈)和i-t长期(100 h)稳定性(图3f-i)。此外,羟基-界面水质子交换策略具有广泛的通用性。对于Ru-M(OH)x/MF(M = Fe、NiFe、Cu)和M-Ni(OH)2/NF(M = Ir、Pt),电化学活化后其碱性HER性能普遍得到改善。![]()
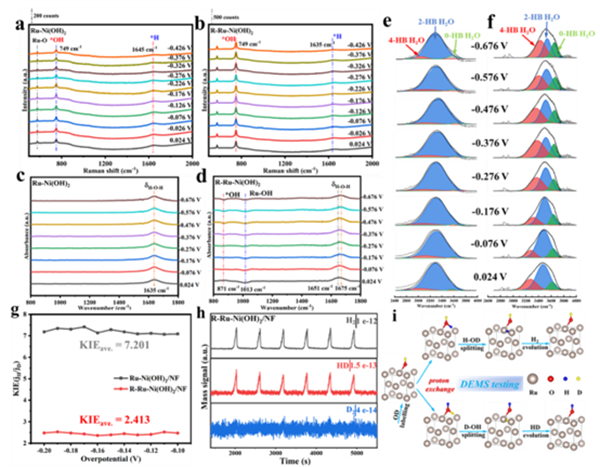
图4 (a) Ru-Ni(OH)2和(b) R-Ru-Ni(OH)2在1 M KOH溶液中从0.024至-0.426 V vs.RHE的原位拉曼光谱。(c, e) Ru-Ni(OH)2和 (d, f) R-Ru-Ni(OH)2在从0.024至-0.676 V vs.RHE的原位衰减全反射表面增强红外吸收光谱(ATR-FTIR)。(g) R-Ru-Ni(OH)2和Ru-Ni(OH)2的动力学同位素效应(KIE)值。(h) R-Ru-Ni(OH)2在1 M KOD/D2O溶液中经CV活化后,通过原位微分电化学质谱(DEMS)检测的产氢信号。(i) 氘标记的R-Ru-Ni(OH)2的DEMS测试示意图。
为了揭示R-Ru-Ni(OH)2碱性HER的反应机理与活化羟基的积极作用,采用原位光谱对活化前后Ru-Ni(OH)2的HER过程进行了研究。原位拉曼/红外光谱(图4a-f)证明,R-Ru-Ni(OH)2拥有更多的表面羟基参与反应,改善了OH中间体演变动力学,有利于界面水的质子交换过程(包括吸附与解离)。R-Ru-Ni(OH)2计算的KIE值更低(图4g),表明HER中O-H更容易断裂以及水解离的加速。DEMS测试结果分析表明,在含D的电解液标记之后,R-Ru-Ni(OH)2出现了明显的HD信号。HD主要来源于KOD/D2O电解液中电化学活化标记的OD物种,通过质子交换与之后的HDO裂解-脱附,从而形成HD气体。这些有力地证明了稳定的表面活化羟基能够显著参与OH-H2O质子交换反应,促进HER动力学(图4h,i)。 ![]()
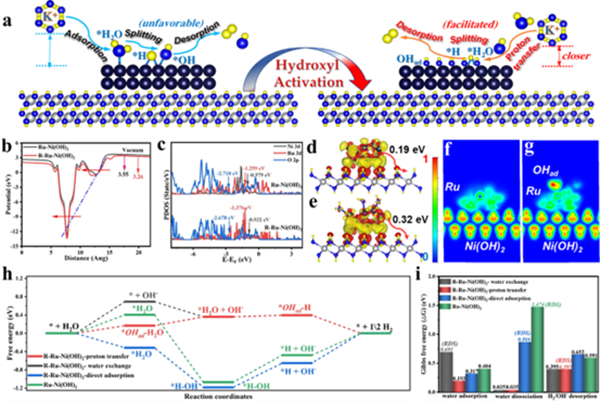
图5 (a) Ru-Ni(OH)2在HER过程中活化前后电子转移及反应中间体演变路径的示意图。(b) R-Ru-Ni(OH)2和Ru-Ni(OH)2的功函数。(c) R-Ru-Ni(OH)2和Ru-Ni(OH)2中Ru、Ni、O原子的部分态密度(PDOS)。(d) Ru-Ni(OH)2和 (e) R-Ru-Ni(OH)2的Bader电荷(DCD)。(f) Ru-Ni(OH)2和 (g) R-Ru-Ni(OH)2的电子局域函数(ELF)。 (h) R-Ru-Ni(OH)2以及Ru-Ni(OH)2中Ru位点在不同路径下的HER反应的吉布斯自由能变化。 (i) R-Ru-Ni(OH)2和Ru-Ni(OH)2中Ru位点在不同路径下的水吸附、水解离和氢/羟基解吸的能垒。
图5a展示了Ru-Ni(OH)2核壳催化剂在羟基活化前后及HER过程中电子/中间体的转移/演变路径。以Ru-OHad为反应位点,可实现更低能垒的质子交换过程,从而促进液态水转移到催化剂表面,同时在羟基位点存在的条件下加速水解离与*H脱附进程。通过WF、PDOS、DCD、ELF以及吉布斯自由能计算,证明了活化OHad位点的电子/能带结构得到改善(图5b-i)。表面活化的OHad位点直接参与并优化了OH-H2O质子交换反应,实现了水吸附并加速了后续水解离过程。同时,由于额外OHad位点的存在改善了*H脱附行为,显著降低了决速步骤反应能垒,极大地提高了催化剂的反应活性。
总结与展望
总之,通过采用简单的电化学活化策略成功将羟基物种引入到Ru-Ni(OH)2核壳催化剂的Ru表面,并验证了引入的活化羟基物种能直接促进碱性HER反应。获得的R-Ru-Ni(OH)2催化剂展现出优异的碱性HER性能。原位光谱与质谱等分析证实了活化后的R-Ru-Ni(OH)2催化剂存在足够多的羟基物种,并直接参与析氢反应;同时,作为优异的质子受体促进了水解离与*H脱附进程。DFT计算进一步验证了羟基物种的形成改善了Ru活性位点的电子转移能力,并优化了反应路径与决速步骤;同时,Ru-OHad与界面水直接质子交换形成*H2O物种,裂解脱附后形成H2,从而极大促进了HER动力学。此外,该电化学激活策略亦表现出普适性。
文献信息
https://doi.org/10.1021/acscatal.4c07602